CLUSTAL Tutorial
Clustal Omega / ClustalW : Multiple alignment of nucleic acid and protein sequences
ClustalW is the oldest of the currently most widely used programs for multiple sequence alignment. Clustal Omega is the latest version of CLUSTAL series. ClustalO is faster and more accurate because of new HMM alignment engine.
In this tutorial, we explain some of the features of the CLUSTAL web application.
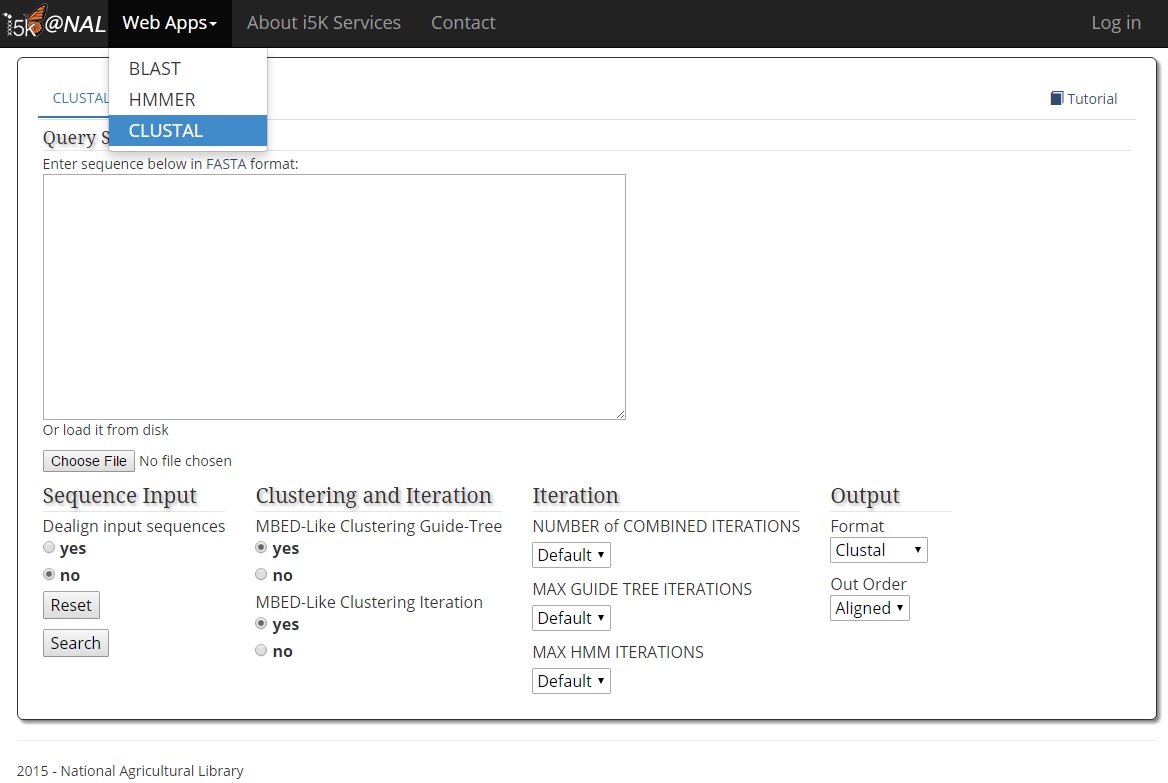
To begin, navigate to the CLUSTAL web application via the 'Web Apps->CLUSTAL' menu bar, or go directly to http://i5k.nal.usda.gov/webapp/clustal/.
We offer both ClustalW and Clustal Omega. Both programs perform multiple sequence alignments. Because the two programs have completely different parameter setting, please refer to the program manuals for details.
Clustal Omega Manual: http://www.clustal.org/omega/README
ClustalW Manual: http://www.clustal.org/download/clustalw_help.txt
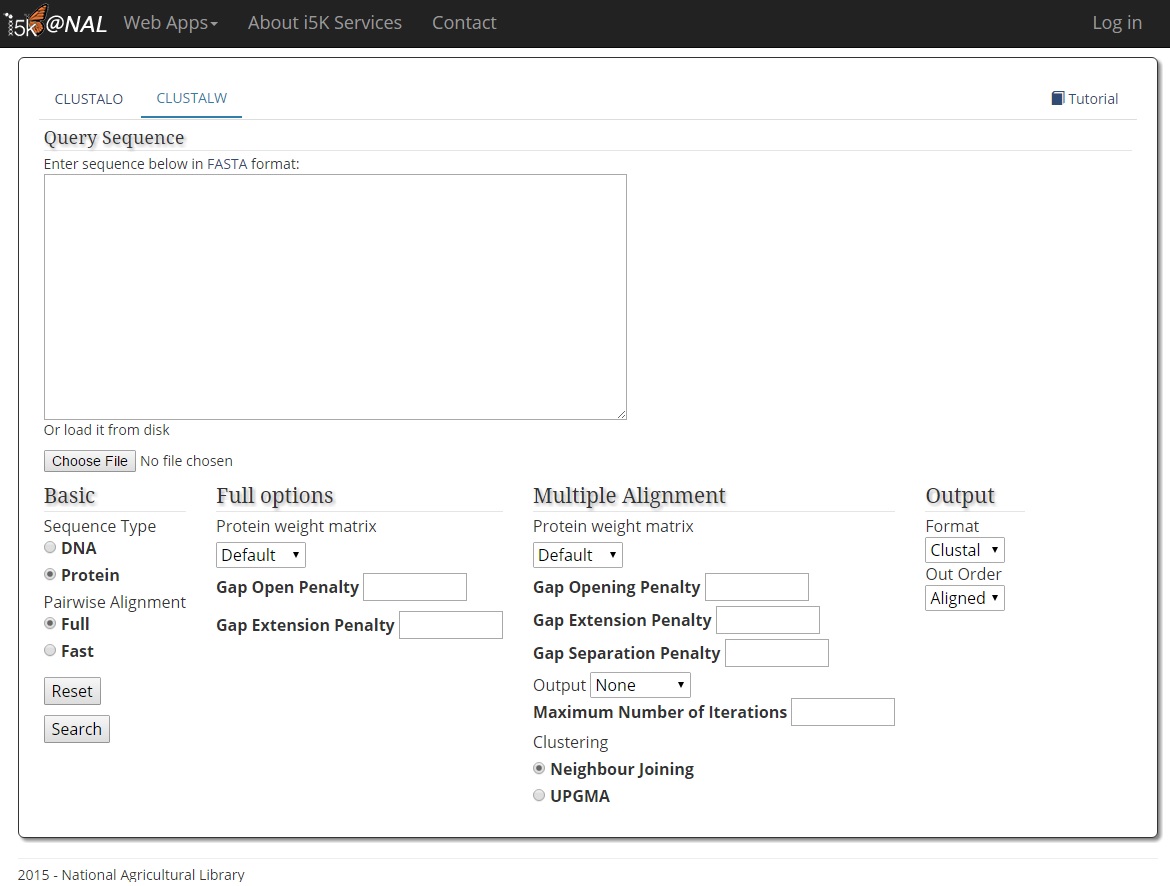
Paste your query sequence into the 'Query Sequence' field, or load it from disk.
The CLUSTAL program only accepts the fasta format as query input. Once the correct CLUSTAL program is selected, you can customize the parameters for your search. Hit the 'Search' button when finished.

A page will display indicating that your query is running. You can store the indicated query link if you wish to come back to your CLUSTAL results at a later time. Results are stored for one week.

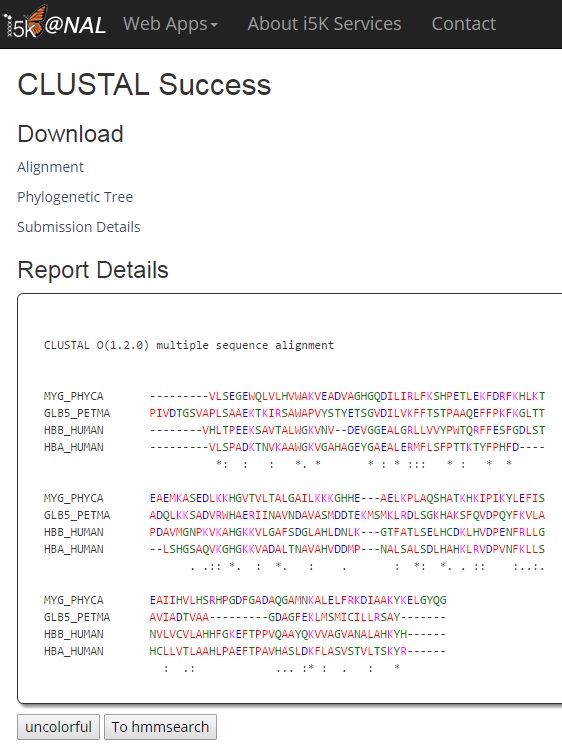
When the CLUSTAL program has finished, your results will be displayed.
In 'Download', you can download the alignment file (shown in 'Report Details', phylogenetic tree (if available) and submission details including parameters used in this job.
In 'Report Details', the 'Colorful' button makes alignment for easier visualization. The 'To Hmmsearch' button redirects users to hmmsearch using CLUSTAL result as input for hmmsearch.